| | The FDA is engaged in numerous activities to protect and promote public health during the COVID-19 pandemic. For CDER, these efforts include accelerating development of treatments for COVID-19, maintaining and securing drug supply chains, providing guidance to stakeholders, advising developers on how to handle clinical trial issues, and keeping the public informed. Information on some of CDER's efforts related specifically to drugs and COVID-19 can be found here in the 2020 issues of the newsletter. Recent updates are provided below: The FDA's Clinical Methodologies Group within CDER's Office of Medical Policy received a $9.2 million grant through the HHS Office of the Assistant Secretary for Planning and Evaluation's (ASPE) Patient Centered Outcomes Research Trust Fund. The grant will fund expansion of the CURE ID platform to allow automated data collection from electronic health records (EHR) worldwide and clinical disease registries for COVID-19 and other difficult-to-treat infectious diseases. COVID-19 EMERGENCY USE AUTHORIZATIONS AND UPDATES - May 21, 2021: The FDA updated the definition for patients who may be considered at high risk for COVID-19 to include additional medical conditions and factors associated with increased risk for progression to severe disease. This update applies to the emergency use authorizations (EUAs) for REGEN-COV (Casirivimab and Imdevimab) and Bamlanivimab and Etesevimab. More information is available in the fact sheets for each EUA:
- June 3, 2021: The FDA reissued the Letter of Authorization for REGEN-COV (Casirivimab and Imdevimab) treatment for COVID-19 to authorize:
- Dosage changes from 1200 mg of casirivimab and 1200 mg of imdevimab to 600 mg of casirivimab and 600 mg of imdevimab;
- A new coformulation presentation that contains 600 mg of casirivimab and 600 mg of imdevimab in a single vial, and
- Addition of subcutaneous (under-the-skin) injection as an alternative route of administration when intravenous (administered into a vein) infusion is not feasible and would lead to delay in treatment.
- July 30, 2021 Update: FDA revised the authorization for REGEN-COV monoclonal antibody therapy for post-exposure prophylaxis (prevention) for COVID-19 to add an authorization of REGEN-COV for emergency use as post-exposure prophylaxis (prevention) for COVID-19 in adults and pediatric individuals (12 years of age and older weighing at least 40 kilograms) who are at high risk for progression to severe COVID-19, including hospitalization or death. REGEN-COV also remains authorized for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
- May 26, 2021: FDA issues emergency use authorization for Sotrovimab for treatment of COVID-19. Sotrovimab is a monoclonal antibody that is specifically directed against the spike protein of SARS-CoV-2 and is authorized for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kilograms) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
- June 24, 2021: FDA issues emergency use authorization for Tocilizumab Monoclonal Antibody for Treatment of COVID-19. Tocilizumab is a monoclonal antibody that reduces inflammation by blocking the interleukin-6 (IL-6) receptor and is authorized for the treatment of COVID-19 in hospitalized adults and pediatric patients (2 years of age and older) who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
- July 28, 2021: FDA revises the EUA for baricitinib now authorizing baricitinib alone for the treatment of COVID-19 in hospitalized adults and pediatric patients two years of age or older requiring supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO).
COVID-19 INVESTIGATIONAL DRUGS-FDA UPDATES/SUMMARIES/STATEMENTS - May 17, 2021: FDA provided summary information about the status of CytoDyn, Inc.'s development program for the monoclonal antibody investigational drug, leronlimab, for the treatment of COVID-19. The data currently available from recent CytoDyn clinical trials do not support the clinical benefit of leronlimab for the treatment of COVID-19.
- May 26, 2021: Important update about bamlanivimab/etesevimab with regard to the P.1 and the B.1.351 variants in multiple states (from HHS/ASPR and FDA).
- June 25, 2021 Update from HHS/ASPR and FDA: The Assistant Secretary for Preparedness and Response (ASPR) is immediately pausing all distribution of bamlanivimab and etesevimab together and etesevimab alone (to pair with existing supply of bamlanivimab at a facility for use under EUA 094) on a national basis until further notice. In addition, FDA recommends that health care providers nationwide use alternative authorized monoclonal antibody therapies, and not use bamlanivimab and etesevimab administered together at this time.
COVID-19 RELATED GUIDANCES | | SPOTLIGHT ON CDER SCIENCE | | | Ensuring the Rigor of Regulatory Science: CDER Conducts Laboratory and Clinical Studies to Investigate Reports of NDMA Production from Ingested Ranitidine Products  Concerns that ranitidine, a popular drug to treat peptic ulcer and gastroenterological reflux disease, could readily be converted to the carcinogen NDMA led FDA to request market withdrawal. As part of its investigation, CDER needed to develop new analytical methods for NDMA detection in ranitidine drug products. These improved methods indicated that actual levels were much lower than previously reported. To further address the possibility that ranitidine could be converted to NDMA in people who take the drug, CDER conducted a randomized clinical trial, the results of which concluded that ranitidine does not increase NDMA under physiological conditions. Based in part on this research, FDA may consider allowing ranitidine-containing products back on the market if they are proven to be stable, with low, acceptable amounts of NDMA that do not increase during storage. Learn more. | A First-in-Kind Pediatric Electrocardiogram Data Warehouse for Prevention of Sudden Cardiac Death in the Young The causes of sudden cardiac death in the young (SCDY) are not well understood, and children who die of it often do not have a known condition that puts them at risk. The FDA is collaborating with partners in academia and industry to create a pediatric Electrocardiogram Data Warehouse as a resource for prevention of SCDY. The data warehouse will contain ECG's and other data collected in a standardized format at public screening events. Development of this first-in-kind pediatric ECG data warehouse is expected to advance development of clinical guidelines for assessing risk in children, evaluation of drug safety, and biomarkers for use in pediatric clinical trials. Learn more. | |  | | | REGULATORY SCIENCE IMPACT STORIES | | | CDER is continuing to highlight its regulatory science research in a series of regulatory science impact stories. Recent posts include:  Advancing our understanding of how consumers and physicians perceive and react to drug advertising Although FDA does not approve prescription drug advertising or other promotional materials, the reviewers in CDER's Office of Prescription Drug Promotion (OPDP) continually monitor prescription drug advertising and promotional labeling to help ensure that it is neither false nor misleading, follow up on complaints about alleged violations of the law and regulations, and initiate compliance actions against purveyors of prescription drugs sold with deceptive claims. Using a variety of approaches derived from the social and communication sciences, OPDP also leads a diverse and dynamic research program to investigate how patients and health care providers perceive and interpret promotional materials and ultimately respond to them. Learn more. |  A physiologically based pharmacokinetic model to support dosing recommendations for antidepressants used during pregnancy Failure to treat major depressive disorder during pregnancy may endanger the health of both mother and child. However, safety, efficacy, and dosing information for drugs administered during pregnancy are scarce and many expectant mothers are still hesitant to take necessary medications due to a lack of reassuring clinical safety information. CDER's Office of New Drugs has worked with the National Center for Toxicological Research (NCTR) research scientists to develop a physiologically based pharmacokinetic (PBPK) model for pregnancy that can predict exposures to the antidepressant drug sertraline. The pregnancy PBPK model was converted into a prototype web-based interactive dosing tool and is being further developed into a software application that clinicians could use to predict the dose adjustments needed to maintain a effective levels of the drug as pregnancy progresses. Learn more. |  The Impact of Continuous Manufacturing Processes on the Viral Safety of Therapeutic Proteins Despite its promise for increasing the efficiency with which therapeutic proteins can be manufactured, continuous manufacturing creates unique challenges for removal of any retrovirus-like particles that may be produced in the cells used to express the protein. CDER researchers and collaborators have recently developed systems and models which can allow manufacturers to carefully assess the efficiency with which viruses are removed during continuous manufacturing. Learn more. | 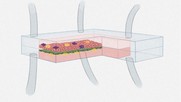 Three-dimensional (3D) cell culture platforms as drug development tools Advances in microengineering and stem cell technologies have led to the development of innovative platforms in which tissue-specific cells are cultured in three dimensions to replicate the physiology of human organs and tissues. These microphysiological systems have the potential to improve the efficiency of drug development by decreasing reliance on costly animal testing and increasing our capacity to predict the efficacy and potential toxicities of new drug candidates. The Division of Applied Regulatory Science in CDER is investigating the performance of these systems and the contexts of use in which they can advance drug development. Learn more. | | | | Recent Scientific Publications by FDA/CDER Staff can be found here. Recent publications include the following. Physiologically- based pharmacokinetic modeling to support determination of bioequivalence for dermatological drug products: scientific and regulatory considerations CDER researchers discuss the scientific and regulatory considerations for the development, verification, and validation of dermal PBPK models used to help establish bioequivalence for topically applied dermatologic products and the challenges posed by the lack of local bioavailability data for these products. Learn more. Visualizing topical drug uptake with conventional fluorescence microscopy and deep learning CDER-led research demonstrates how the combination of standard epi-fluorescence imaging with deep learning can be used to the visualize and quantify fluorescent drugs in human skin. Learn more. Subgroup analyses in oncology trials: regulatory considerations and case examples CDER reviewer/researchers summarize key issues related to subgroup analyses in the benefit-risk assessment of cancer drugs and provide case examples to illustrate approaches that the FDA Oncology Center of Excellence (OCE) has taken when considering the appropriate patient population for cancer drug approval. Learn more. Effect of formulation and peptide folding on the fibrillar aggregation, gelation, and oxidation of a therapeutic peptide CDER researchers report on their experimental and computation-based investigations of a therapeutic peptide. These studies may provide insights into formulation strategies to reduce physical and chemical degradation of peptide products with defined conformations and help ensure drug quality. Learn more. | | | For more information, please visit the FDA In Brief Webpage. Scientific Internships and Fellowships / Trainees and Non-U.S. Citizens Whether you're an undergraduate looking to pursue a career in science, a graduate student seeking experience in regulatory science, a postgraduate looking for fellowship opportunities, or a senior scientist pursuing research experience in your field of expertise, FDA offers you many paths to learning about the exciting field of regulatory science. Click here for more information. | | | | |



No comments:
Post a Comment